ИСПОЛЬЗОВАНИЕ МЕТОДА ПОЛНОГЕНОМНОГО АССОЦИАТИВНОГО ИССЛЕДОВАНИЯ В СЕЛЕКЦИИ СВИНЕЙ
ИСПОЛЬЗОВАНИЕ МЕТОДА ПОЛНОГЕНОМНОГО АССОЦИАТИВНОГО ИССЛЕДОВАНИЯ В СЕЛЕКЦИИ СВИНЕЙ

Аннотация
Разработка геномных технологий, направленных на повышение эффекта селекции в животноводстве, напрямую зависит от правильных методических подходов и применения актуальных достоверных программ. Одним из таких направлений является геномная оценка по экономически значимым хозяйственно-полезным признакам. В данной статье описан расчет геномных оценок племенной ценности и проведение полногеномного ассоциативного исследования, на примере среднесуточного прироста свиней породы дюрок. Проанализированы выявленные гены, имеющие биологическую направленность, и приведен анализ их обогащения. Результаты данных исследований можно использовать для написания научных статей, учебных пособий и на курсах повышения квалификации.
1. Введение
Ускорение генетического улучшения основных хозяйственно-полезных признаков, имеющих значение в секторе экономики, путем применения селекции с помощью маркеров повышает эффективность отбора и разведения свиней. Однако анализ генетической архитектуры этих признаков остается сложной и трудной задачей, и для любого признака у свиней было выявлено несколько определенных причинных вариантов – это миссенс-мутация гена рецептора меланокортина 4 (MC4R), которая участвует в энергетическом гомеостазе и соматическом росте , а также в эффективности корма у свиней . Аналогичным образом, миссенс-мутация в гене протеинкиназы AMP-активируемой некаталитической субъединицы гамма 3 (PRKAG), расположенном на хромосоме 5 Sus scrofa, и, как было показано, влияет на уровень гликогена в мышцах у породы гемпшир . Его эффект настолько велик, что данная аллель была устранена из многих коммерческих линий свиней, основанных на гемпшире, с помощью селекции. Было также показано, что мутация сплайсинга в гене каталитической цепи фосфорилазы В киназы гамма (PHKG1), расположенном на хромосоме 3 генома свиней, влияет на содержание гликогена . Чтобы продемонстрировать, что варианты являются причинными, требуются дальнейшие биологические анализы с использованием новых программ. Однако вычислительные методы, такие как полногеномное исследования ассоциаций (GWAS), являются важным первым шагом в идентификации генов-кандидатов. Целью GWAS по одному маркеру является выявление базовой причинной генетической основы признака путем независимого тестирования каждого генетического варианта на статистическую связь с интересующим признаком. Полногеномное секвенирование многих животных является дорогостоящим, и поэтому панели полиморфизма одного нуклеотида (SNP) средней или высокой плотности, в настоящее время, все чаще используются для GWAS у сельскохозяйственных животных. Данный метод основан на неравновесии сцепления (LD) между SNP, присутствующими на панели, и причинными вариантами для идентификации геномных областей, связанных с признаком. Однако высокая LD распространяется на большие расстояния в популяциях скота, и идентификация причинного варианта из множества вариантов, которые могут находиться в высокой LD с SNP GWAS, остается сложной задачей.
Геномная селекция, основанная на прогнозировании племенной ценности (геномной племенной ценности или GEBV) животных с учетом геномной информации, меняет стратегии и подходы к разведению молочного КРС и ряда других видов животных. Отправными точками для применения геномной селекции у свиней стали разработка первой коммерческой панели однонуклеотидного полиморфизма для высокопроизводительного генотипирования, секвенирование генома свиньи и применение статистических и методологических подходов, впервые разработанных для молочного скота , а затем адаптированных к особенностям свиноводческой отрасли .
В связи с вышеизложенной актуальностью, целью данного исследования являлся подробное описание методического подхода применения метода GWAS, полученного на рассчитанных геномных оценках племенной ценности среднесуточного прироста свиней коммерческой породы дюрок.
2. Методы и принципы исследования
Материалом исследования являлись данные геномных оценок племенной ценности показателя среднесуточного прироста 408 хряках породы дюрок 2017-2020 годов рождения, проходивших откорм на автоматических кормовых станциях. В тексте встречается следующее сокращение признака среднесуточного прироста – ADG, г.
Геномный BLUP (GenomicBLUP, GBLUP) также идентичен по применяемой методологии решения уравнения обычному BLUP, однако матрица родства A заменяется матрицей геномного сходства G, учитывающей как генотип по каждому SNP, так и частоту этого генотипа:
С – «центрированная матрица» SNP-маркеров:
C = M - 2P, где
M – матрица содержания SNP-файла (закодированная зиготность SNP-маркеров, где 0 – гомозиготность AA, 1 – гетерозиготность AB, 2 – гомозиготность BB), P – матрица частот генотипов, где Pi=2(pi-0,5); pi – частота встречаемости минорного аллеля (MAF) для i-го SNP.
C’ – транспонированная матрица C.
Таким образом, между животными рассчитывается совершенно иной тип родства, позволяющий также учесть, какие сегменты хромосом унаследовали потомки от предков.
Исходя из этого, решение уравнения GBLUP будет иметь вид:
G-1 – обратная матрица геномного сходства.
Оценки племенной ценности, получаемые в результате применения GBLUP, называют GEBV (Genomic EBV).
Проведение GWAS основано на определении корреляции между генетическими маркерами средней (несколько десятков тысяч) или высокой (несколько сотен тысяч) плотности и сложными количественными признаками. С этой целью находит применение R-пакет GenABEL, в котором реализована общая линейная смешанная модель. Модель включает в себя случайный полигенный эффект, при котором матрица дисперсии-ковариации пропорциональна единице по всему геному . Для выполнения GWAS предварительно проводят оценку племенной ценности животных, используя смешанные математические модели. В качестве примера можно привести следующую:
где y – наблюдаемое значение признака;
µ – популяционная константа X – фиксированный эффект, учитывающий группу содержания, год и сезон;
w – живая масса индивида, рассматриваемая, как коварианта;
c – совокупный эффект SNP;
a – вектор случайных аддитивных генетических эффектов при σ_α^2 ~ N(0, Gσ_α^2), где G – матрица геномного сходства особей, вычисленная на основании данных генотипирования с использованием ДНК-чипов средней или высокой плотности, а σ_α^2 – дисперсия аддитивных полигенных эффектов;
K – коэффициент регрессии оцениваемого признака на живую массу индивидов;
e – остаток модели при σ_e^2 ~ N(0, Iσ_e^2), где I – единичная диагональная матрица, σe2 – остаточная дисперсия.
В данном исследовании GWA-анализ проводили с использованием ДНК-чипа Porcine GGP HD (платформа GeneSeek Genomic Profiler, «Neogene», США), содержащим ~70 тыс. SNP. Контроль качества и фильтрацию данных генотипирования для каждого SNP и каждого образца выполняли с использованием программного пакета PLINK 1.9, применяя следующие фильтры:
- call-rate по всем исследуемым SNP для индивидуального образца не ниже 90% (--mind 0,10);
- call-rate для каждого из исследованных SNP по всем генотипированным образцам не ниже 90% (--geno 0,10);
- частота встречаемости минорных аллелей (MAF) ≥0,05 (--maf 0,05);
- отклонение генотипов по SNP от распределения по Харди-Вайнбергу в совокупности протестированных образцов (--hwe 1e-3).
Достоверность результатов GWAS оценивается по P-значению, которое подвергается коррекции на множественное тестирование. Пороговое значение может быть разным в разных исследованиях, но чаще всего для анализа, в котором рассматривается десятки и сотни тысяч SNP, оно принимается равным 5×10-8. Для корректировки результирующего значения P используются методы проверки нескольких гипотез, такие как Бонферрони, Бенджамина-Хохберга или расчет ожидаемой доли ложных отклонений (FDR)
.В результате проведенного анализа, для проведения анализа ассоциаций были отобраны 40 069 SNP, прошедшие фильтрацию по всем параметрам качества.
Для выявления ассоциаций SNP маркеров с изучаемыми признаками применяли регрессионный анализ, реализованный в PLINK 1.90 (--assoc --adjust --qt-means). Для подтверждения достоверного влияния SNP и определения значимых регионов в геноме свиней использовали тест для проверки нулевых гипотез по Бонферрони при пороговом значении p <1,25×10-5, 0,05/40069. Данные визуализировали в пакете qqman с помощью языка программирования R.
Поскольку стратификация популяции сильно влияет на надежность GWAS, анализ участка квантиль-квантильности (Q-Q) считается эффективным способом определения надежности результатов GWAS. В графике Q-Q горизонтальная ось представляет ожидаемое значение -log10P, а вертикальная ось представляет наблюдаемое значение -log10P. Диагональная линия представляет зависимость вида y=x, показывает 95% доверительный интервал, основанный на бета-распределении. Общее отклонение над диагональной линией идентичности обычно указывает на сильную стратификацию популяции. Отклонения от диагональной линии указывают на то, что предполагаемое распределение неверно или что образец содержит значения, возникающие каким-либо другим образом, аналогичные тем, которые порождаются истинным объединением. График Q-Q строится с использованием средств визуализации языка программирования R.
Для поиска генов-кандидатов, локализованных в области идентифицированных SNP, использовали геномный ресурс Sscrofa11.1. Функциональные аннотации генов выполняли с привлечением веб-программы PANTHER 19.0, базы данных DAVID и Pig QTL data base. Чтобы получить представление о функциональном обогащении генов, были впоследствии проведены анализы путей обогащения термина Gene Ontology (GO)
.3. Основные результаты
Впервые в России апробация GWAS анализа по хозяйственно-полезным признакам свиней породы дюрок произошла в 2019 г. и продолжается улучшаться по сей день, включая новые подходы и методы исследования полногеномного анализа , , . В данной научной работе скомпонован комплекс подходов к получению достоверных генов для дальнейшего их использования в геномной селекции свиней.
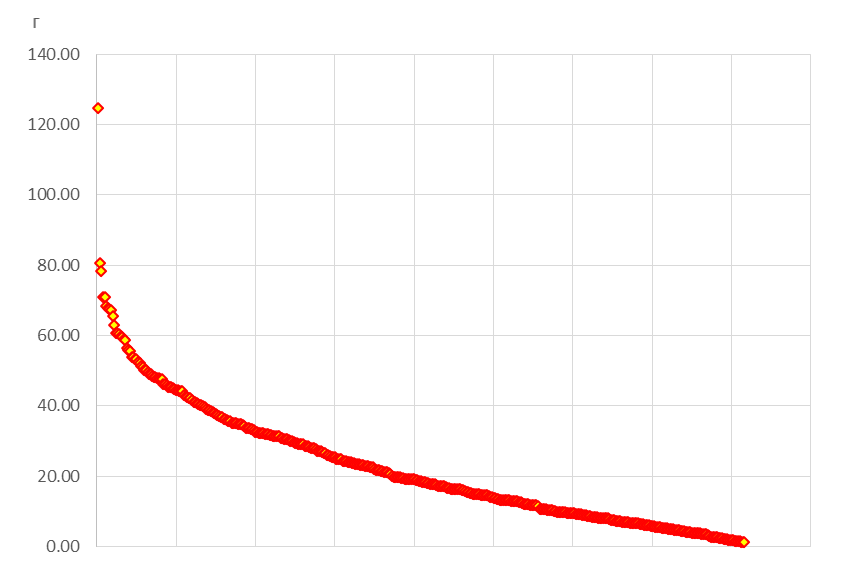
Диаграмма распределения лучших геномных оценок племенной ценности по показателю среднесуточного прироста хряков породы дюрок
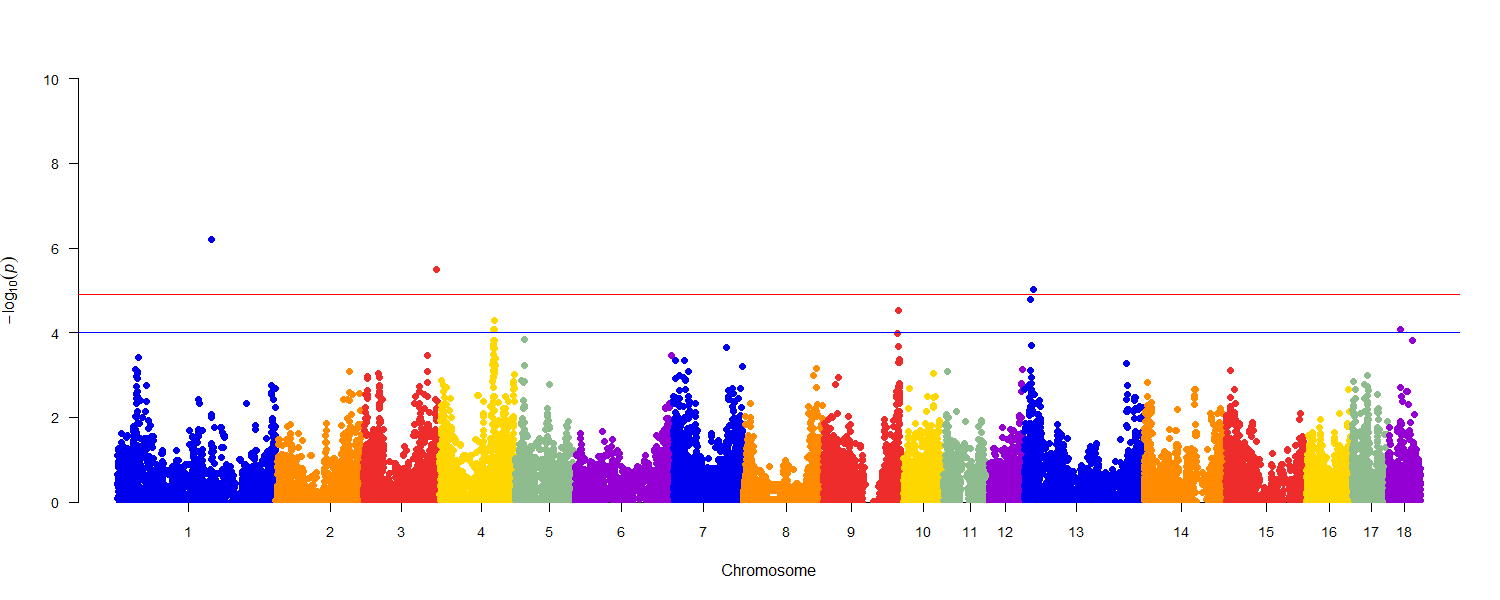
Распределение однонуклеотидных мутаций по хромосомам свиней породы дюрок в связи с уровнем достоверности (-log10 (p) по вероятностному суггестивному значению (синяя линия, p<1,25×10-4) и критерию Бонферрони (красная линия, p<1,25×10-5) для GEBV среднесуточного прироста
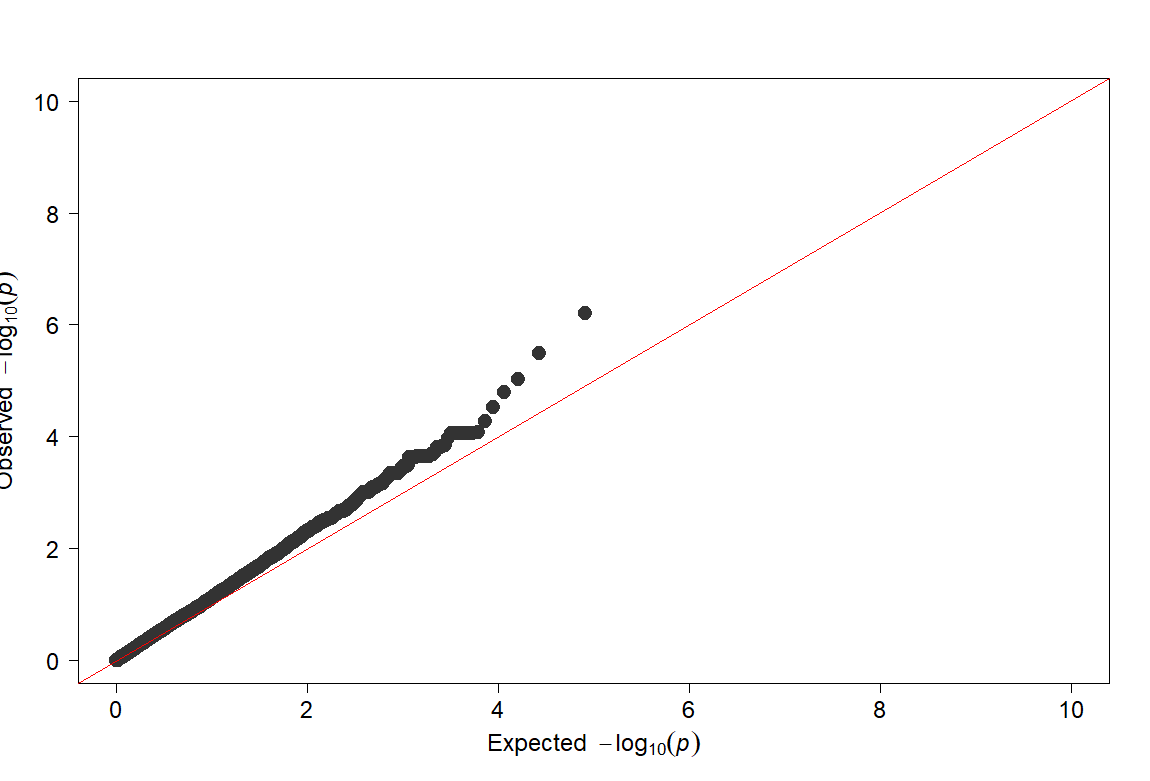
Квартиль вероятностного распределения ожидаемого и наблюдаемого отклонений от нормального распределения для значений достоверности
Наибольшее количество генов расположены на 4 хромосоме (n=60).
Таблица 1 - Структурная аннотация достоверно выявленных SNP по признаку среднесуточного прироста
№хр. | SNPПозиция | Р | ГенПротяженность |
1 | WU_10.2_1_180284104162 607 316
| 6,15E-07 | NEDD4L162364053..162736515 |
3 | ALGA0116500127 464 779 | 3,17E-06 | KIDINS220127382587..127477753 |
4 | ASGA002096992 646 954 | 0,000224 | MBOAT2127243797..127365443 |
ASGA002098392 939 481 | ID2127500759..127503207 | ||
ASGA002098993 062 179 | ETV392964808..92979129 | ||
ASGA002099093 081 498 | ETV3L93005259..93011259 | ||
4_10162428792 954 515 | 0,00019 | ARHGEF1193045533..93160756 | |
ALGA002683593 017 369 | 0,000224 | PEAR193178854..93205186 | |
ALGA002687193 492 857 | NAXE93493234..93495912 | ||
MARC002582692 994 199 | LRRC7193162453..93176013 | ||
WU_10.2_4_10241932793 717 288 | 0,000152 | NTRK193219509..93237944 | |
INSRR93239678..93258075 | |||
PRCC93297859..93325499 | |||
HDGF93341081..93351809 | |||
METTL25B93358140..93366169 | |||
ISG20L293366188..93372407 | |||
MRPL2493352997..93357805 | |||
SH2D2A93282383..93293214 | |||
CRABP293388198..93393861 | |||
NES93412660..93421899 | |||
IQGAP393495999..93556915 | |||
HAPLN293462306..93468394 | |||
BCAN93432331..93450895 | |||
GPATCH493486365..93492837 | |||
TTC2493500900..93512039 | |||
MEF2D93581563..93616980 | |||
ALGA002693194786666 | 8,55E-05 | GBA194583905..94606689 | |
ALGA00269379 4908711 | 8,55E-05 | MUC194626317..94631194 | |
M1GA000613694920625 | 8,55E-05 | MTX194606576..94611439 | |
M1GA000613994952966 | 8,55E-05 | TRIM4694632073..94642756 | |
MARC004704394995753 | 5,27E-05 | THBS394612702..94623905 | |
MARC005106194385015 | 8,55E-05 | KRTCAP294643300..94646436 | |
MARC009309394339347 | 8,55E-05 | SLC50A194663057..94665486 | |
EFNA194666457..94673442 | |||
DPM394661568..94662223 | |||
EFNA394709941..94719350 | |||
DCST194748430..94768322 | |||
ADAM1594736639..94748093 | |||
EFNA494730790..94735578 | |||
PMVK94861500..94913743 | |||
PBXIP194837365..94847652 | |||
FLAD194807280..94813512 | |||
SHC194818226..94831766 | |||
DCST294768533..94781482 | |||
PYGO294832279..94836548 | |||
LENEP94805409..94807175 | |||
CKS1B94815769..94819583 | |||
DBWU000096096207274 | 0,000156 | CHTOP95996030..96006511 | |
S100A196007026..96012305 | |||
GATAD2B95766989..95864502 | |||
INTS395904072..95944805 | |||
SLC27A395898780..95903599 | |||
S100A1396007280..96025120 | |||
SNAPIN95987524..95989527 | |||
S100A1696030930..96038602 | |||
NPR195957220..95974253 | |||
ILF295980862..95987568 | |||
S100A1496027746..96029878 | |||
S100A596093204..96096870 | |||
S100A296073496..96076825 | |||
S100A496088102..96091160 | |||
S100A396084049..96086829 | |||
S100A696098264..96099686 | |||
S100A796176718..96178694 | |||
5 | H3GA001585617163494 | 0,000142 | SLC4A816842667..16921669 |
SCN8A16977468..17173831 | |||
9 | ASGA0044888129535988 | 3,02E-05 | PTPN14129114668..129248898 |
DRGA0009891128247578 | 0,000106 | PLA2G4A127853581..128164825 | |
WU_10.2_9_142385342129688707 | 0,00021 | SMYD2129263524..129316226 | |
PROX1129527704..129583693 | |||
13 | ALGA006793210415894 | 1,64E-05 | UBE2E29998097..10380564 |
ASGA005636915586736 | 9,42E-06 | RBMS314968921..16458470 | |
ALGA009741821938080 | 8,36E-05 | GRM820945049..21756952 | |
WU_10.2_18_4622045842107509 | 0,0001513 | ITPRID141491840..41768828 | |
NEUROD641757356..41763837 | |||
ADCYAP1R141915180..41972327 | |||
GHRHR42030505..42046178 | |||
AQP142063482..42076741 | |||
MINDY442096351..42202511 |
Примечание: жирным шрифтом выделены гены, внутри которых локализован выявленный SNP; №хр. – номер хромосомы; SNP – однонуклеотидный полиморфизм; Р – достоверность выявленного SNP
Далее все выявленные гены были соотнесены с биологическими функциями в веб-программе PANTHER 19.0 (рис. 3). Идентифицировано 11 групп генов GO, включая метаболический процесс, который из всех представляет наибольший интерес с сопряженностью со среднесуточным приростом.
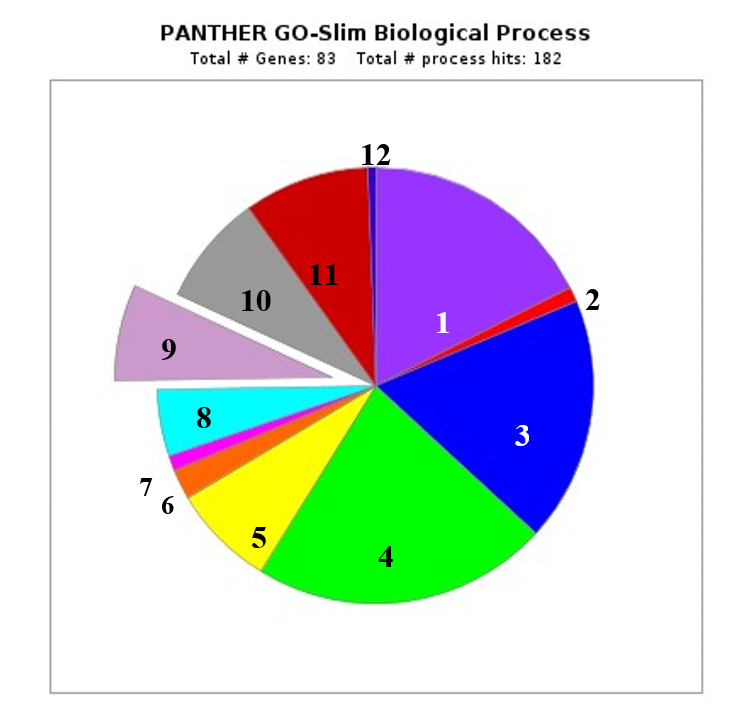
Распределение идентифицированных генов по биологическим функциям (GO):
1 - категория не присвоена; 2 - биологический процесс, участвующий в межвидовом взаимодействии организмов (GO:0044419); 3 - биологическая регуляция (GO:0065007); 4 - клеточный процесс (GO:0009987); 5 - процесс развития (GO:0032502); 6 - гомеостатический процесс (GO:0042592); 7 - процесс иммунной системы (GO:0002376); 8 - локализация (GO:0051179); 9 - метаболический процесс (GO:0008152); 10 - многоклеточный организменный процесс (GO:0032501); 11 - ответ на стимул (GO:0050896); 12 - ритмический процесс (GO:0048511)
выделенная часть в диаграмме – метаболический процесс (GO:0008152)
Таблица 2 - Функциональная аннотация генов, ассоциированных с выявленными SNP
№ хр. | Ген | DAVID (GOTERM_BP) | Pig QTL |
1 | NEDD4L | нет | QTL:169088 |
3 | KIDINS220 | сигнальный путь фактора роста нервов | нет |
MBOAT2 | модификация липидов | нет | |
ID2 | регуляция процессов липидного обмена, двигательный ритм, развитие альвеол молочной железы | нет | |
4 | NAXE | транспорт липидов, регулирование оттока холестерина | нет |
NTRK1 | развитие многоклеточного организма, развитие нервной системы | нет | |
INSRR | развитие многоклеточного организма, определение мужского пола | нет | |
CRABP2 | транспорт жирных кислот, эмбриональный морфогенез передних конечностей | нет | |
HAPLN2 | развитие костной системы, развитие центральной нервной системы | нет | |
BCAN | развитие костной системы, центральной нервной системы | нет | |
MEF2D | развитие сердца | нет | |
GBA1 | метаболический процесс, морфогенез мозга, нервно-мышечный процесс | нет | |
THBS3 | развитие хрящевой пластинки, окостенение | нет | |
DCST1 | слияние сперматозоида с плазматической мембраной яйцеклетки, участвующей в однократном оплодотворении, распознавание сперматозоидов и яйцеклеток | нет | |
ADAM15 | врожденный иммунный ответ, переход от эпителия сердца к мезенхиме, иммунный ответ на опухолевую клетку, | нет | |
PBXIP1 | кроветворение, развитие суставного хряща | нет | |
SHC1 | ангиогенез, развитие сердца | нет | |
PYGO2 | развитие почек | нет | |
S100A1 | регуляция сердечных сокращений | нет | |
SLC27A3 | процесс метаболизма длинноцепочечных жирных кислот | нет | |
S100A14 | реакция на липополисахарид | нет | |
S100A6 | клеточный ответ на вирус | нет | |
SCN8A | нет | QTL:160641; QTL:160642 | |
9 | PTPN14 | нет | нет |
PLA2G4A | процесс метаболизма глицерина, процесс метаболизма арахидоновой кислоты | нет | |
PROX1 | развитие почек, развитие легких | QTL:179339 | |
13 | RBMS3 | защитный ответ на опухолевую клетку | QTL:23467 |
18 | GRM8 | нет | QTL:19443; QTL:264227 |
NEUROD6 | развитие нервной системы, развитие органов чувств | нет | |
GHRHR | нет | QTL:1186; QTL:1192; QTL:1191 |
Примечание: жирным шрифтом выделены гены, внутри которых локализован выявленный SNP
Анализ обогащения показал наибольшую локализацию генов по кластеру 1 (коэффициент обогащения = 6,88), принадлежащего к функциям: связывание ионов кальция – 12 генов, Р=4,1х10-5; кальций-зависимое связывание с белками – 10 генов, Р=2,1х10-12 и перинуклеарная область цитоплазмы – 8 генов, Р=9,4х10-4.
В группу связывания ионов кальция (GO:0005509) входят гены: S100A2, S100A16, S100A1, S100A6, S100A13, PLA2G4A, S100A5, S100A4, S100A14, S100A3, S100A7, THBS3. В кальций-зависимое связывание с белками (GO:0048306) – гены S100A2, S100A16, S100A1, S100A6, S100A13, S100A5, S100A4, S100A14, S100A3, S100A7. И в группу, отвечающую за перинуклеарную область цитоплазмы (GO:0048471) – гены SNAPIN, S100A16, S100A1, S100A6, S100A13, S100A4, S100A14, THBS3.
4. Заключение
Данное научно-практическое исследование можно использовать как методические аспекты структуризации расчета геномной оценки в области свиноводства, применяя актуальные программы для получения достоверного результата.
По выявленным генам, внутри которых локализованы SNP, а именно, NEDD4L, KIDINS220, NAXE, S100A1, PLA2G4A и GRM8, в дальнейшем будут разработаны тест-системы и проведен массовый скрининг на остальных коммерческих породах свиней.
Результаты данных исследований можно использовать для написания научных статей, учебных пособий и на курсах повышения квалификации.